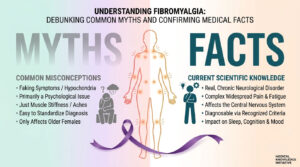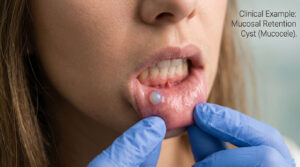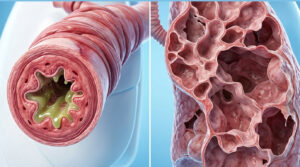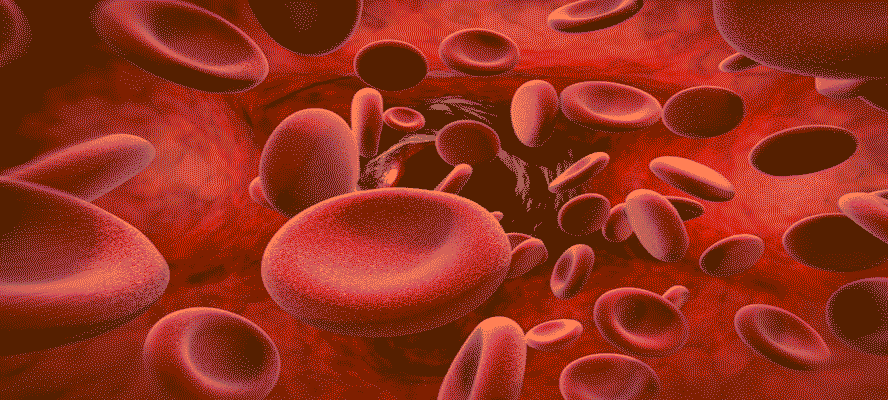
Ancient struggles over skin colour, geographical whereabouts and appearance evoked a raging discrimination among the races. For quiet a few decades superiority and inferiority were based on the colour of an individual and sidetracked were the medical conditions, which might have afterall been responsible for acquiring certain irrevocable disorders. In the chest of those secrets, which we have to unlock is an unorthodox scientific story that talks more about us ‘Humans’! So is it only the colour that differentiates human beings into Africans, East Asians and Caucasians to name a few? What role do our phenotype and genotype together play, when it comes to these fatalities? How authentic is the big fat risk for hemoglobin disorders across different races?