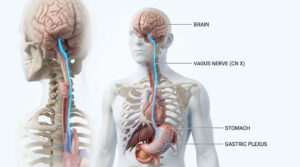
Present in the African population for thousands of years and known by many local names, Sickle cell disease’ was first described in the year 1910, by a cardiologist Dr. James B. Herrick in a dental student of African origin named Walter Clement Noel. He presented with painful episodes and symptoms of anemia along with pulmonary symptoms, his blood smear also showed peculiarities in Red blood cells (RBCS) having a shape of a sickle