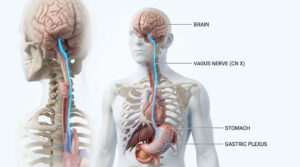
in a ward of a hospital, a child was standing at the window, grasping the bars and sadly watching the children of his age playing and running around happily in the playground. It was a situation that could have filled anybody’s heart with compassion. The child was there for blood transfusions. Because he inherited from his parents a deadly disorder known as Thalassemia.