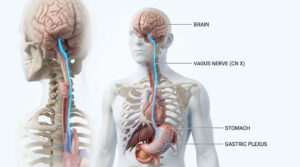
Who is at risk for thalassemia?
Thalassemia is a purely genetic disorder that is carried over in generations either as carriers or as diseased individuals. Below mentioned are the factors which are the reasons for individuals who are prone
to the disease at a higher level:
Family history of thalassemia:
Thalassemia is passed from parents to children through mutated hemoglobin genes. If you have a family history of thalassemia, you may have an increased risk of the condition.
Certain ancestry:
Thalassemia occurs most often in people of Italian, Greek, Middle Eastern, Asian and African ancestry.