Most people file Huntington’s disease under “brain disorder” and stop there. That label is accurate, yet it hides half of what the disease does to the body.
Table of Contents
The muscles waste. Weight drops even when meals stay the same. Swallowing turns risky in ways that can be fatal. These physical changes sit close to the center of how Huntington’s unfolds, not at the edges.
Skeletal muscle makes up close to 40% of body weight, so a disease that reaches into muscle reshapes the whole body, not just movement. Reading the muscle side correctly is one of the most practical things a patient or caregiver can do.
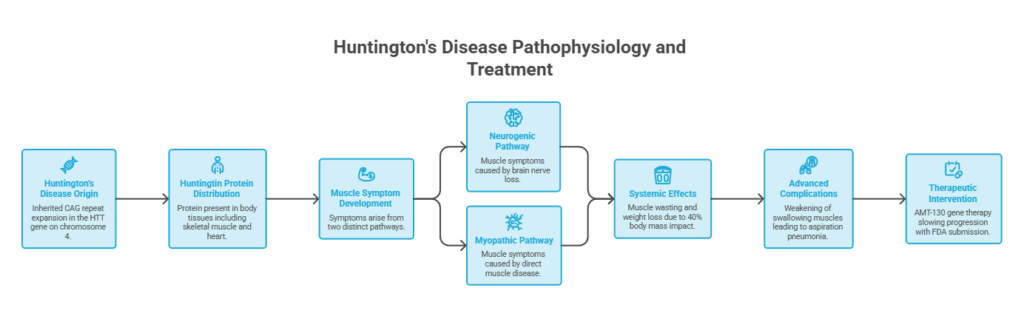
Quick answer: Yes, Huntington’s disease affects muscle as well as the brain. It acts through two routes: nerve loss in the brain that scrambles movement control, and the mutant huntingtin protein working directly inside skeletal muscle. Together these cause involuntary movements, weakness, muscle wasting, and unexplained weight loss. Spotting the muscle signs early helps families protect strength, nutrition, and swallowing safety.
At a Glance
- Huntington’s disease is inherited, caused by a CAG repeat expansion in the HTT gene on chromosome 4.
- The huntingtin protein sits in tissues across the body, including skeletal muscle and the heart, not the brain alone.
- Muscle symptoms arise two ways: from brain nerve loss (neurogenic) and from direct muscle disease (myopathic).
- Muscle wasting and weight loss are common, partly because muscle is about 40% of body mass.
- Swallowing muscles weaken as the disease advances, and aspiration pneumonia is the leading cause of death.
- A gene therapy called AMT-130 has reported slowing progression, with an FDA submission underway.
What Huntington’s Disease Actually Is (and Why “Brain Disease” Is Only Half the Story)
Huntington’s disease (HD) is an inherited disorder that damages the brain and body over roughly 15 to 20 years. Symptoms usually begin between ages 30 and 50, according to the National Institute of Neurological Disorders and Stroke.

The disease is progressive and, for now, fatal. Changes build slowly, drifting from subtle shifts in movement and mood toward heavy dependence in daily life.
Patients booking tests with us often ask whether HD is “a nerve problem or a muscle problem.” The honest answer is both, and the reason sits inside the gene that causes it.
The HTT gene, CAG repeats, and the huntingtin protein
HD comes from a mutation in the HTT gene on chromosome 4. That gene carries a repeated three-letter DNA sequence, C-A-G, that gets copied too many times in people who develop the disease.
A repeat count of 40 or more leads to HD within a person’s lifetime. Counts of 36 to 39 land in a “reduced penetrance” range, where symptoms may or may not appear.
Larger repeat counts tend to bring earlier and more severe disease, a pattern Mayo Clinic laboratories describe in their HD testing overview. The disease also shows genetic anticipation, meaning repeat sizes can grow as the gene passes to the next generation.
The gene builds a protein called huntingtin. When the CAG stretch runs too long, that protein misfolds, clumps, and turns toxic to the cells carrying it.
Each child of a parent with HD has a 50% chance of inheriting the faulty gene, as the American Brain Foundation notes. That inheritance pattern is why HD moves through family trees across generations.
Why the protein reaches far beyond the brain
This is the fact that reframes the disease. Huntingtin is not brain-only. It is expressed widely across the body, including the heart, kidneys, liver, and skeletal muscle.
The brain shows the highest levels, which is why thinking and mood symptoms stand out. But the same toxic protein sits inside muscle fibers, doing quiet damage there too.
Our medical reviewers note that this whole-body distribution is why HD symptoms feel so scattered. A disease driven by one faulty protein in many tissues will never stay boxed inside a single organ.
HD is rare, though likely more common than older counts suggested. Roughly 1 in 10,000 Americans lives with the disease, and modern insurance-claims data point to diagnostic rates reaching 15 per 100,000 people. The Huntington’s Disease Society of America serves as a central US resource for families navigating those numbers.
How Huntington’s Disease Affects Your Muscles
Muscle trouble in HD arrives through two separate doors. Grasping both is the key to reading symptoms correctly and acting in time.
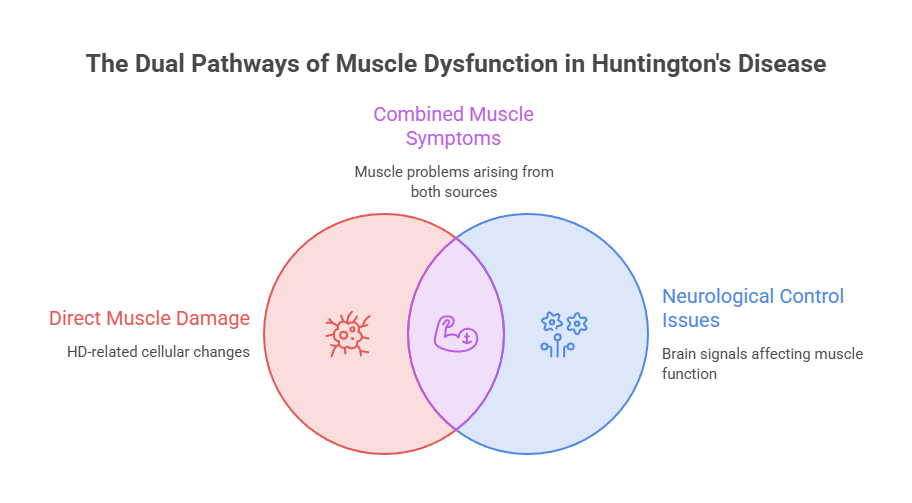
The brain route (neurogenic)
The classic story is neurogenic, meaning it starts with nerve loss. HD destroys neurons in the striatum and cerebral cortex, the brain regions that plan and smooth out movement.
As those control centers fail, muscles get scrambled signals. The result is involuntary movement, stiffness, and lost coordination that no effort of will can override.
This route drives chorea, the dance-like, fidgety movements many people link with HD. It also produces dystonia and rigidity as the disease moves forward.
Motor symptoms often begin in the distal extremities, the fingers and toes and small facial muscles, before spreading to the rest of the body. That outward creep is a hallmark clinicians watch for.
The muscle route (myopathic)
The newer story is myopathic, meaning the trouble starts inside the muscle itself. Research shows that mutant huntingtin inside skeletal muscle disrupts how those fibers work, apart from any brain damage.
Studies summarized by Medical News Today point to disruption of a chloride channel protein called ClC-1, which helps regulate muscle excitability. When chloride and calcium signaling go wrong, muscles fire and relax abnormally.
Laboratory work also finds mitochondrial dysfunction, lower ATP (the fuel muscle cells burn), oxidative stress, and disturbed calcium handling in HD muscle. A review in the National Library of Medicine describes skeletal muscle wasting as a recognized hallmark of the disease.
One mouse study found the muscle’s internal tubule network stayed intact, but individual tubules narrowed, disrupting the calcium signaling behind strength and stamina. That kind of defect helps explain the weakness and fatigue patients describe.
Why researchers now call HD “a myopathy as well as a neurodegenerative disease”
A team at Wright State University stated it plainly. Their work in the Journal of General Physiology, summarized by the university, found muscle maturation is disrupted in HD models.
The researchers argue HD behaves as a muscle disorder layered on top of a brain disorder. Some muscle damage may even develop independently of the nerve loss.
That distinction is not hair-splitting. It opens a path to treatments aimed at muscle directly, and it hints that muscle changes could work as easier markers to track the disease than repeated brain scans.
| Symptom or Feature | Brain (neurogenic) origin | Muscle (myopathic) origin | What patients and caregivers notice |
| Involuntary movements (chorea) | Nerve loss in the striatum disrupts movement control | Altered muscle excitability from ClC-1 and calcium changes | Fidgety, dance-like motions of the hands, face, and trunk |
| Stiffness (dystonia and rigidity) | Faulty signaling in basal ganglia pathways | Impaired muscle contraction and relaxation | Twisted postures, cramped limbs, a stiff neck |
| Weakness and fatigue | Reduced drive from the motor cortex | Mitochondrial dysfunction and low ATP in fibers | Tiring fast, weak grip, dropping objects |
| Muscle wasting (atrophy) | Lower activity and denervation over time | Direct fiber degeneration and protein-quality defects | Shrinking arms and legs, visible thinning |
| Weight loss | Appetite and behavior changes | Muscle is about 40% of body mass, so loss drops weight | Falling weight despite normal eating |
| Swallowing trouble (dysphagia) | Loss of coordination in throat nerve control | Weakness in tongue and throat muscles | Coughing at meals, choking, slow eating |
How Muscle Symptoms Change Across the Stages of HD
Muscle symptoms don’t stay still. They shift in character as the disease moves through early, middle, and late stages, and knowing the arc helps families plan rather than react.
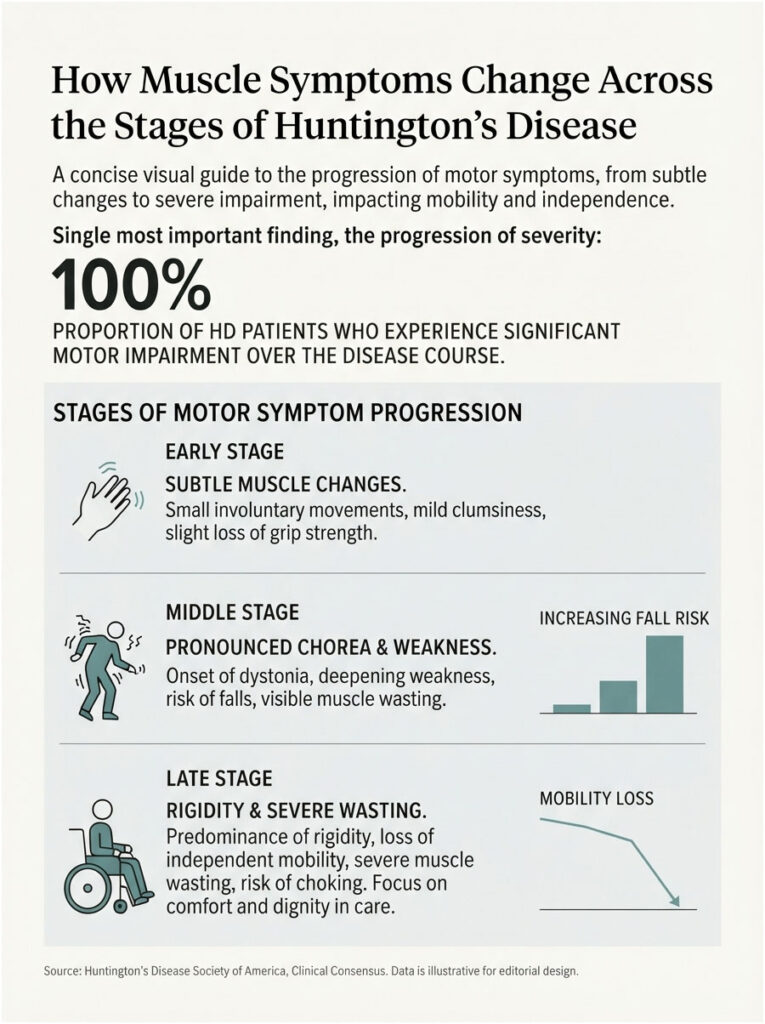
Early stage
In early HD, muscle changes are easy to miss or misread. Small involuntary movements, mild clumsiness, and a slight loss of grip strength can look like stress or ordinary aging.
Chorea often shows up first as restlessness, a fidget in the fingers or face. Weakness and fatigue may appear before anyone connects them to HD.
Remarkably, physical changes can trail brain changes that started far earlier. NINDS notes that adults carrying the mutation show measurable brain structure changes up to 20 years before a clinical diagnosis. Early muscle signs deserve a neurologist’s eye, not a wait-and-see.
Middle stage
In the middle stage, movement symptoms become harder to ignore. Chorea grows more pronounced, and dystonia begins pulling the body into sustained, sometimes painful postures.
Weakness deepens, balance falters, and falls become a real risk. Weight loss and visible muscle wasting often set in here, even when appetite holds.
Across the families we serve, this is frequently the stage when the physical toll finally prompts a full care plan. Physical therapy and nutrition support matter most when started before a crisis, not after.
Late stage
In late HD, rigidity and bradykinesia (slowed movement) tend to overtake chorea, which often fades. Movement becomes effortful, and many patients lose independent mobility.
Swallowing muscles weaken further, raising the danger of choking and aspiration. Muscle wasting and weight loss can be severe, and full-time care is usually needed.
Our medical reviewers note that late-stage care leans heavily on comfort, safety, and dignity. Skilled support for swallowing, nutrition, and movement remains valuable at every point.
The Muscle Symptoms People Miss
Families often fix their attention on memory and mood, then get blindsided by physical decline. Knowing the muscle signs early gives everyone more room to act.
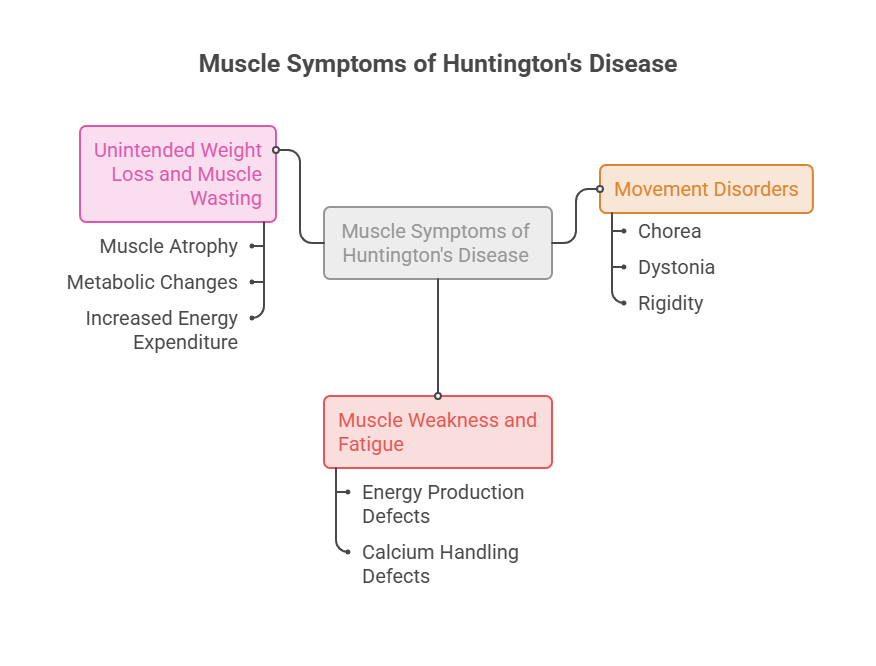
Chorea, dystonia, and rigidity
Chorea is the most familiar sign: brief, irregular, involuntary movements that flow from one muscle group to the next. Early on, it reads like ordinary fidgeting.
Dystonia is different. It involves sustained contractions that twist the body into awkward, sometimes painful positions, and it tends to worsen as HD advances.
Rigidity and bradykinesia become more disabling later. As one movement-disorder specialist told the American Medical Association, the illness can feel like several neurologic diseases stacked at once.
Muscle weakness and fatigue
Weakness in HD is easy to write off as “getting older” or “being tired.” That mistake costs families time.
The muscle-level defects in energy production and calcium handling leave fibers less able to generate force. Grip weakens, stamina drops, and everyday tasks start to demand real effort.
In cases reviewed by our medical team, early weakness is often logged as unrelated to HD until a clear pattern forms. Keeping simple notes on when and how weakness appears helps a neurologist connect the dots.
Unintended weight loss and muscle wasting
Weight loss is one of the most telling and most overlooked features of HD. People can shed pounds steadily even while eating a normal or larger diet.
Muscle wasting, or atrophy, is a major driver. As fibers degenerate, muscle mass shrinks, and total body weight falls with it.
Why weight drops even when eating normally
The math is simple once you see it. Skeletal muscle accounts for roughly 40% of total body weight, according to reviews in the National Library of Medicine.
Lose muscle, and you lose a large share of body mass, no matter what’s on the plate. Metabolic changes and the extra energy burned by constant movement widen the deficit.
Across the families we serve, unexplained weight loss is frequently the symptom that finally triggers a full workup. It deserves attention, not reassurance.
When Muscle Trouble Turns Dangerous: Swallowing and the Heart
Some muscle effects are merely inconvenient. Others threaten life. Two systems demand special watchfulness: the swallowing muscles and the heart.
Dysphagia and aspiration pneumonia
Swallowing is a muscular act, run by nerves and dozens of small muscles in the mouth and throat. HD disrupts both the timing and the strength of that process.
The problem, called dysphagia, grows more common as the disease advances. A fiberoptic swallowing study in the National Library of Medicine found dysphagia in 35% of early-stage, 94% of moderate-stage, and 100% of advanced-stage patients.
When swallowing fails, food or liquid can slip into the airway instead of the stomach. That misdirection, called aspiration, can trigger a serious lung infection known as aspiration pneumonia.
Aspiration pneumonia is the leading cause of death in HD. Medical News Today reports that older research tied up to 87% of HD deaths to it.
How swallowing muscles fail as HD progresses
Early on, the changes can be silent. A person may swallow slightly out of sync without obvious choking, so caregivers miss it.
Later, coughing at meals, throat-clearing, a wet-sounding voice, and outright choking appear. Eating too fast or not chewing enough makes the risk worse.
Our medical reviewers note that a swallow evaluation with a speech-language pathologist should happen early, not after a scare. Small changes to food texture and meal pace can prevent a hospital trip.
HD and the heart muscle
The heart is a muscle too, and HD does not spare it. Patients face higher rates of irregular rhythms and heart-muscle dysfunction.
Cardiovascular disease is a leading cause of death in HD, second to aspiration pneumonia, and accounts for a meaningful share of cases. Palpitations, fainting, or breathlessness should always go to a physician.
Our lab partners report that heart symptoms in HD get less attention than the movement and cognitive signs. Screening and monitoring matter here too.
HD Muscle Symptoms vs Look-Alike Conditions
HD muscle symptoms can mimic other diseases, which is one reason diagnosis sometimes drags. Chorea, weakness, and wasting appear in several disorders.
HD differs from Parkinson’s disease, which centers on rigidity and slowness with a resting tremor. It differs from ALS, a pure motor-neuron disease without the cognitive and psychiatric profile of HD.
It also differs from primary myopathies, where muscle disease occurs without the brain and behavioral changes. The mix of movement, thinking, and mood symptoms, plus a family history, points toward HD, confirmed by a genetic test through providers such as Mayo Clinic.
| Statistic | Figure | Source |
| Americans living with manifest HD | ~21,000 to 40,000 | Neurology (2020 analysis) |
| Approximate population rate | ~1 in 10,000 | American Brain Foundation |
| Diagnostic frequency (US claims data) | 6.5 per 100,000, up to ~15 per 100,000 | Neurology / US claims studies |
| Typical age symptoms begin | 30 to 50 years | NINDS |
| Dysphagia rate by disease stage | 35% early, 94% moderate, 100% advanced | Swallowing study, NLM |
| AMT-130 gene therapy result | ~75% slowing at high dose over 3 years | uniQure / CNN |
Managing Muscle Symptoms and Protecting Function
There’s no cure yet, but muscle symptoms can be managed and function protected for longer. A team approach works best, and starting early beats catching up later.
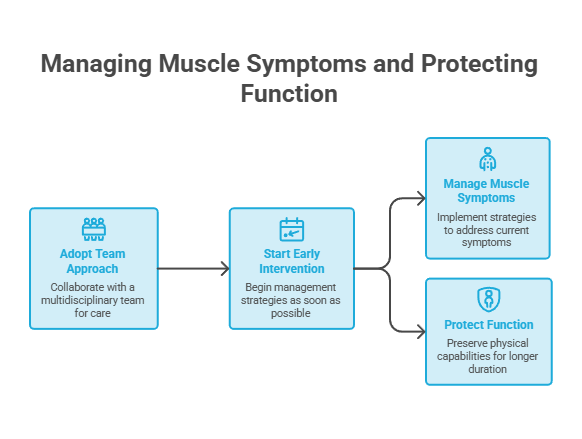
Build a multidisciplinary team
No single clinician handles HD alone. The strongest care pulls together a neurologist, physical therapist, speech-language pathologist, dietitian, and mental-health support.
HDSA designates Centers of Excellence across the US that coordinate this kind of team care. Asking a neurologist for a referral to one can simplify a hard road.
Patients commonly ask us for a single “muscle pill” for HD. That does not exist; management blends medication, therapy, nutrition, and support around the individual.
Movement-symptom medications
Doctors can treat chorea with prescription medications, including a class known as VMAT2 inhibitors. These aim to calm involuntary movements, not to slow the disease.
Every drug carries trade-offs, and the right choice depends on the mix of movement, mood, and other health issues. This is a decision for a neurologist who knows the full picture, since HealthCareOnTime does not provide medical treatment.
Physical therapy, strength work, and safe exercise
Movement helps. Reviews of exercise in HD, including one in the National Library of Medicine, describe physical activity as a useful tool to support motor function and quality of life.
A physical therapist can design a safe program that builds strength, protects balance, and lowers fall risk. Stopping movement to “save energy” tends to backfire, speeding muscle loss.
The aim is steady, supervised activity matched to the person’s stage. Even late in the disease, guided movement supports circulation, mood, and function.
Nutrition, calorie needs, and swallowing safety
Because HD burns through calories and wastes muscle, nutrition is frontline care, not an afterthought. Many patients need calorie-dense meals to hold their weight.
A registered dietitian can build a plan that meets higher energy needs while keeping food safe to swallow. Texture changes, thickened liquids, and slower meals cut choking and aspiration risk.
Regular weight checks catch decline early. A falling number on the scale is a signal to adjust the plan, not to wait and watch.
What the new gene therapy does (and doesn’t do) for muscle
The biggest recent news is AMT-130, an experimental gene therapy from uniQure. Preliminary results reported that a high dose slowed disease progression by about 75% over three years, as covered by CNN and Science.
This is a one-time treatment delivered by brain surgery, and it works by lowering huntingtin in the brain. The company has moved toward an FDA submission, with plans laid out on its pipeline page.
AMT-130 targets the brain; muscle-directed approaches are a separate frontier
Here’s an honest caveat. AMT-130 lowers the toxic protein in the brain, so its main effect falls on the neurologic side of the disease.
Muscle-directed therapies, aimed at the myopathic damage inside fibers, remain a separate research frontier. The muscle-as-marker idea, using measurable muscle defects to track progression, is also still developing.
Results so far come from a small group and rely on comparison controls rather than a large trial, so caution is warranted. Even so, this is the first time any treatment has shown meaningful slowing of HD, which shifts the outlook for families.
| What you notice | What it may signal | Recommended action |
| New grip weakness or frequent dropping | Muscle weakness or an early motor change | Log examples with dates; ask a neurologist for a movement exam |
| Unplanned weight loss over weeks | Muscle wasting or rising calorie needs | Track weekly weight; request a dietitian referral and a calorie-dense plan |
| Coughing, throat-clearing, or choking at meals | Swallowing-muscle involvement (dysphagia) | Ask for a swallow evaluation with speech-language pathology promptly |
| More falls or unsteadiness | Rigidity, weakness, or coordination loss | Add physical therapy; make the home safer against falls |
| Palpitations, fainting, or breathlessness | Possible heart-muscle involvement | Report to a physician; ask about cardiac screening such as an ECG |
| At risk with no symptoms yet | Premanifest status | Discuss genetic counseling and baseline monitoring before symptoms start |
Common Mistakes Families Make with HD Muscle Symptoms
Small missteps add up over a long disease. A few patterns show up again and again.
Blaming everything on the brain
When families treat HD as a pure brain disease, they watch memory and mood while missing weight loss, weakness, and swallowing risk. Those physical signs need equal attention.
Reading symptoms through both lenses, brain and muscle, leads to better and earlier decisions. It also tells caregivers which specialist to call first.
Cutting back on movement
The instinct to rest and preserve energy is natural, but it speeds muscle loss. Inactivity feeds atrophy, and atrophy feeds weakness.
Supervised exercise, tuned to the person’s stage, holds onto strength and function longer. Movement is medicine here, within the limits a therapist sets.
Waiting too long for therapy
Speech and physical therapy often get called in only after a crisis, such as a choking episode or a bad fall. Earlier is far better.
Bringing these specialists in at the first hint of swallowing change or balance trouble buys time and safety. Our medical reviewers note that proactive referrals consistently improve day-to-day life for HD families.
Frequently Asked Questions
Does Huntington’s disease cause muscle weakness?
Yes. Weakness in HD comes from both brain nerve loss and direct muscle disease, where the huntingtin protein disrupts energy production and calcium signaling in fibers. People notice weaker grip, faster fatigue, and difficulty with tasks that once felt easy, often years before advanced symptoms appear.
Why do Huntington’s patients lose weight even when eating normally?
Muscle makes up roughly 40% of body weight, so muscle wasting alone lowers the number on the scale. HD also raises energy demands and shifts metabolism, creating a calorie deficit despite normal or increased eating. Steady, unexplained weight loss is a common and important sign of the disease.
Is muscle wasting in Huntington’s disease reversible?
There’s no proven way to reverse HD-related muscle wasting yet. However, supervised exercise, strength work, and calorie-dense nutrition can slow the loss and preserve function. Stopping activity tends to speed wasting, so guided movement and dietitian support stay central to management at every stage.
Can exercise help muscle symptoms in Huntington’s disease?
Yes. Research reviews describe physical activity as a useful tool for supporting motor function and quality of life in HD. A physical therapist can build a safe plan that maintains strength, protects balance, and reduces fall risk. Consistency matters more than intensity, and programs should match the person’s stage.
What is the difference between chorea and dystonia in HD?
Chorea is brief, irregular, involuntary movement that flows between muscle groups, often looking like restlessness early on. Dystonia is a sustained contraction that twists the body into awkward, sometimes painful postures. Chorea tends to dominate early stages, while dystonia and rigidity increase as HD progresses.
Does Huntington’s disease affect the heart muscle?
Yes. The huntingtin protein is present in heart tissue, and HD patients face higher rates of irregular rhythms and heart-muscle dysfunction. Cardiovascular disease is a leading cause of death in HD after aspiration pneumonia. Palpitations, fainting, or breathlessness should be reported to a physician for evaluation.
Why do people with Huntington’s have trouble swallowing?
Swallowing depends on coordinated throat and tongue muscles controlled by nerves. HD disrupts both their timing and strength, causing dysphagia. This leads to coughing, choking, and the risk of food entering the airway. Because aspiration pneumonia is the top cause of death in HD, swallowing changes need prompt attention.
At what age do muscle symptoms of Huntington’s usually start?
Adult-onset HD typically begins between ages 30 and 50, with muscle and movement symptoms emerging alongside cognitive and mood changes. A rarer juvenile form starts in childhood or adolescence and often features rigidity and slowness rather than chorea. Onset age is influenced partly by the CAG repeat count.
Can a blood or muscle test track Huntington’s progression?
A genetic test confirms HD by measuring CAG repeats, but tracking progression currently relies mainly on clinical exams and brain imaging. Researchers are studying whether measurable muscle defects could serve as easier biomarkers than brain tissue. That approach looks promising but isn’t yet standard clinical practice.
Is Huntington’s disease considered a myopathy?
Increasingly, in part, yes. Researchers argue HD behaves as a myopathy (muscle disease) layered on top of a neurodegenerative disorder, because mutant huntingtin damages muscle fibers directly. Some muscle changes may develop independently of brain nerve loss. This dual nature is now an active and growing area of study.
What medications help HD movement symptoms?
Doctors may prescribe medications, including VMAT2 inhibitors, to reduce chorea, along with other drugs for stiffness or mood. These manage symptoms rather than slowing the disease. Choices depend on the full symptom picture and must be guided by a neurologist, since HealthCareOnTime does not provide medical treatment.
Does the new gene therapy (AMT-130) fix muscle symptoms too?
Not directly. AMT-130 lowers the toxic huntingtin protein in the brain, so its main effect falls on the neurologic side of HD. Muscle-directed therapies remain a separate research frontier. Early results are encouraging but come from small groups, so the treatment’s full impact is still being studied.
Disclaimer: This article is for general education only and does not replace professional medical advice, diagnosis, or treatment. Huntington’s disease needs individualized care from qualified clinicians. Always consult a neurologist or your physician about symptoms, medications, and management. If you notice swallowing difficulty, choking, or rapid weight loss, seek medical guidance promptly.
References
- Huntington’s Disease, National Institute of Neurological Disorders and Stroke
- Overview of Huntington’s Disease, Huntington’s Disease Society of America
- Huntington’s Disease Diagnosis and Treatment, Mayo Clinic
- Huntington’s Disease, Cleveland Clinic
- Huntington’s disease affects muscle as well as brain, Medical News Today
- Researchers reveal Huntington’s disease affects skeletal muscle as well as neurons, Wright State University
- Skeletal muscle pathology in Huntington’s disease, National Library of Medicine
- Molecular mechanisms underlying muscle wasting in Huntington’s disease, National Library of Medicine
- Effects of exercise on skeletal muscle pathophysiology in Huntington’s disease, National Library of Medicine
- Swallowing and dysphagia evaluation in Huntington’s disease, National Library of Medicine
- Prevalence of Huntington’s disease in the US, Neurology
- New Huntington’s disease research, American Brain Foundation
- What doctors want patients to know about Huntington’s disease, American Medical Association
- Experimental gene therapy slows Huntington’s disease progression, CNN
- In a first, a gene therapy seems to slow Huntington disease, Science
- Huntington’s disease programs and pipeline, uniQure